前言
在生命科学研究领域的相关人员,经常使用电子表格来记录和管理实验数据。针对这个问题,我们将会提出一个使用R和Bioconductor管理基因组级实验数据的原则。使用基于R的软件来管理数据似乎并不太常见。通常来说,R仅用于编写程序,构建统计模型。但是,针对复杂生物实验数据的多方面需求这种情况,Bioconductor已经非常着手以统一的方式来详细地注释这些数据。Bioconductor的这种做法可以让我们导入初步分析的结果(例如多个微阵列的扫描结果以及多个测序结果的比对信息),将这些初步的结果与有关实验证的元数据,以及检测的样本信息结合起来,从而将所有的信息都保存在一个R变量中。通过周全的设计该变量的数据结构,程序可以对这些数据进行各种操作,最终获得更加有效的管理和分析环境。
在这一节中,我们会通过一些实验数据来演示一下基本的操作方法,从而,逐步过滤到更加全面的表示方法(
我们将通过对Bioconductor中所表示的实验数据的一些非常基本的表示法,逐步走向更先进的综合表示法,从而理解这种数据操作方法所表现出来的高效率。
案例说明 (Youtube上有相应视频)
1.0 Distinct components from an experiment.
|
|
我们导入了一个GSE5859Subset的实验数据,现在我们使用 setdiff()函数来比较一下导入前后文件的区别,如下所示:
|
|
从结果中我们可以发现,当导入了 GSE5859Subset实验数据中,这个数据中含有3个文件。它们3个分别属于不同的类,如下所示:
|
|
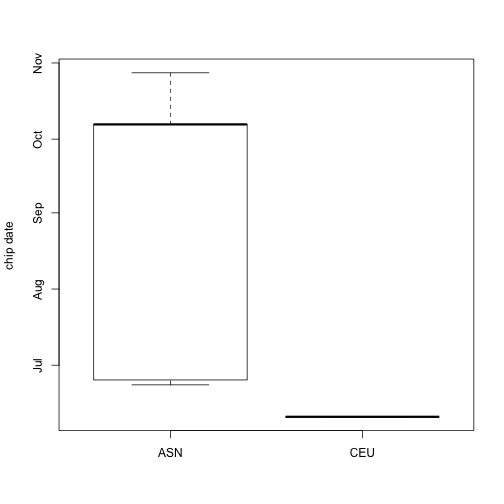
我们可以按照种族绘制绘制出它们不同时间的图形,如下所示:
|
|
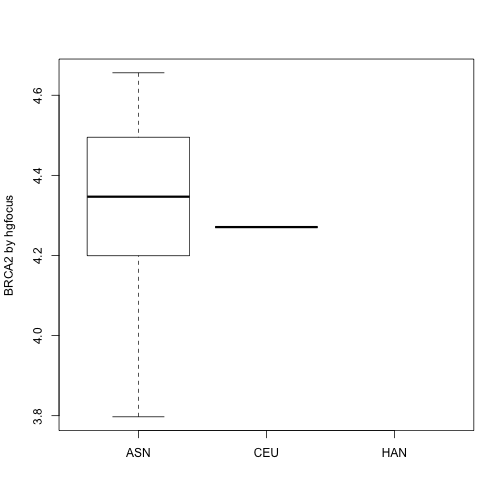
假如我们想查看一下BRCA2在不同种族中的表达情况,我们可以按以下操作进行:
- 我们需要知道
geneExpression这个矩阵中哪一行代表BRCA2的数据,并且我们要知道geneAnnotation哪一列提供了BRCA2的信息(探针名与基因名的对应关系); - 我们需要知道
geneExpression这个矩阵中的顺序与samkpleInfo是一致的,要知道sampleInfo中的信息与geneExpression中的标签的对应关系。
因此,我为了将基因表达矩阵与样本信息联系起来,我们需要对三个数据进行操作(分别为表达矩阵,样本信息,基因注释信息),从而确保这些信息能够匹配起来,如下所示:
|
|
虽然我们绘制出了图形,但是这略微复杂,为了降低操作难度,Bioconductor定义了一个名为ExpressionSet数据结构。
ExpressionSet数据结构
我们使用简单的命令就能构建起ExpressionSet这种数据结构,如下所示:
|
|
上面的es 这个单一变量就浓缩了整个实验的数据,将原来的三个数据变为了一个。现在我们使用es1就能直接绘图,如下所示:
|
|
现在我们已经了解了ExpressionSet容器的设计,你自己也能构建一个ExpressionSet实例。我们将会在下面的实验中,使用一个更加广泛的数据表示方法,如下所示:
|
|
元数据管理(Metadata management)
及时地了解关于实验意图和方法的信息非常有用。GSE5859中使用的一些微阵列数据就是类来源于一篇发表于2004年(PubmedID为15269782)的论文。在联网的情况下,我们可以将论文的信息导入R,如下所示:
|
|
现在查看你R环境中的e变量,使用experimentData(e) <- p后,就能够将PubmedID信息添加到e中。使用experimentData(e)也能发现e中的信息改变了,也含有Pubmed ID信息。
关于ExpressionSet的Why与How
现在我们通过创建以及使用ExpressionSet 实例已经快速了解了这种数据结构的特点。我们现在将要了解一下,为什么要这样设计这种数据结构,以及如何使用这些数据结构。
设计ExpressionSet 这种数据结构的初衷有很多,以下几点最为关键:
- 降低复杂分析,注释以及样本信息编程的难度;
- 支持特征值(其实就是表达值)和样本信息的简单的过滤;
- 在将分析数据传递给分析函数时控制内存占用。
以上就是为什么要设计ExpressionSet这种数据结构。关于如何使用这种结构,这些内容就要涉及一些设计模式问题,在R中,与面向对象编程相关类被称为S4类。
面向对象编程与S4类
面向对象编程(OOP, Object-oriented programming)是计算机编程一个学科,它有助于降低程序的复杂性和维护要求,并能增强程度的互动性。OOP有着各种解决方式,即使在R中,也可以进行OOP,这就是所谓的S4方法。在R中,OOP的基本思想就是一个对象有些特征:
- 将不同类型的数据集中放到一个统一的结构中,
- 它被标记为某个正式指定类的成员,并且可以正式地位于相关对象的系统中,
- 以类特异性方式对泛型方法进行响应。
我们可以使用getClass函数查看一个ExpresionSet的类结构,如下所示:
|
|
从上面我们可以看到,一共有7个组件定义了该类。只有annoataion这个组件占据了一个单独的R类型,也就是character类型。至于其它的组件,都是S4类的一个实例(或者说是更多的R数据类型)。
举例说明一下,MIAME类的定义如下所示:
|
|
因此,我们可以发现,S4可以让我们定义包含各种信息的新数据结。我们虽然可以使用R中非常通用的列表结构来实现这一目的(就是说实现封装不同信息),但是S4允许我们能保证有新结构的属性,以便为这些结构定义的程序不必“检查”它们正在操作什么。这些检查过程内置在了类系统中。
数据框(DataFrame): 表格数据的增强型结构
数据框是R中非常基础的数据结构。Bioconductor中引入了DataFrame类,它可以实现其它一些额外的功能,后面我们会提到。
|
|
show方法描述顶部和底部的行数据,提供有关列数据类型的信息,并且能够比data.frame数据结构更有效地管理列中的复杂数据类所,下面是类定义:
|
|
源于多样本NGS数据的成熟数据表示方法
短读长测序产生了远比微阵列多得多的数据。测序实验也可以通过更加多样化的手段进行分析,因此测序实验能够在不同粒度级别(at various levels of granularity)提供有效的输入。
处理RNA-seq数据的典型流程包括:
- 展示每个周期中碱基所调用的色谱(2008年后就不再提供了);
- 碱基的读取质量值,它们通常以FASTQ格式进行储存;
- 通过基因组或转录本比对后,对各个基因统计的结果;
- 基因组极的多个样本的数据储存,用于推断感兴趣的生物学问题。通常一个样本对应两个FASTQ文件。
Bioconductor中的BAM文件
RNAseqData.HNRNPC.bam.chr14 中含有一些BAM文件,这些BAM文件是8个Hela细胞系样本的双端测序数据,其中4个样本通过RNAi的方式敲低了异质核核糖核蛋白C1和C2(hnRNPC,heterogeneous nuclear ribonucleoproteins C1 and C2)。这个家庭的核蛋白参与pre-mRNA的加工。现在我们演示一下,如何从BAM文件中来统计与HNRNPC有关的读长数目(reads count)。
BAM文件储存在RNAseqData.HNRNPC.bam.chr14 包中,如下所示:
|
|
我们使用Rsamtools包以一个变量的形式管理这些信息,如下所示:
|
|
BAM文件包括关于读长(reads)与基因组比对后的有限数量的元数据。这里我们以BamFileList实例来说明一下:
|
|
我们看到基因”没有指定“,但是我们碰巧知道参考基因组hg19用于进入基因序列的比对。使用[genome(bfl) <- “hg19”可以纠正这点,但是还存在着一个bug。这个bug应该修正一下,以便可以标记使用不同的参考基因组来进行比对或纠正错误,注:这段不太理解,原文如下:
We see that the genome is “unspecified”, but we happen to know that the reference build ‘hg19’ was used to define the genomic sequence for alignment. [genome(bfl) <- “hg19” should rectify this but at present there is a bug. This should be fixed so that attempts to compare alignments using different
references can be flagged or throw errors.]
为了验证是否成功敲除,我们将检查编码HNRNPC的基因组附近的序列的比对情况。稍后,我们将展示如何使用Bioconductor注释工具获取基因地址;不过这个地址仅用于hg19基因组,如下所示:
|
|
这标识了HNRNPC编码基因为hg19注释的染色体14的区域。评估表达式量化的一种简单方法是使用GenomicAlignmentspackage中的summary izeOverlaps。此计数方法将隐式使用多核计算。
上面的代码标识了14号染色体中编码HNRNPC附近的序列。评估基因表达量化的一种简单方法就是使用GenomicAlignmentspackage包中的summarizeOverlaps 函数,这个计算方法隐式地使用了多核计算,如下所示:
|
|
练习
- 检查
hnse中的count组件内容。与HNRNPC敲除相对应的有关量化的指标是什么? - 向
hnse的colData组件中添加一个变量condition,用于指示敲除株与野生株。 - 具有后缀306和307的样本是单个对照的技术重复;308和309同样如此。因此使用了6个不同的生物学样本。现在向
colData中添加一个变量,用于区别这6个样本。
ExpressionSet设计的一些复杂性(进阶)
生物信息学家是在一个不断变化的环境中工作。自Bioconductor建立以来,计算机的容量和通量已经大幅度增加。R语言也性现了很大的变量。在早期的时候,大家都比较注意内存的使用,因为当时在R中,向一个函数传递参数时,会复制内存中的对象, 这种操作比较低效。而“环境”类中的R对象则不会发生复制,这就导致了使用环境来储存大量的数据用于分析。
|
|
“环境”是R中一种非常特殊的对象类型,可以将它视为对表达值矩阵引用的容器。
|
|
练习(可选)
请解释以下运行结果:
|
|
运行结果如下所示:
|
|